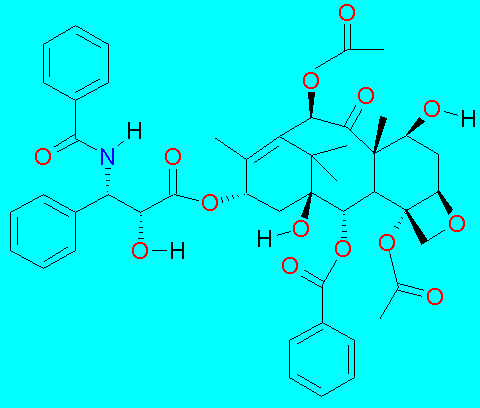
The discovery of its bioactivity was made by a botanist in 1962, Arthur Barclay, under contract to the US NCI (National Cancer Institute), while working for the USDA. Taxol was isolated from the stem and bark extract of Taxus brevifolia by Dr. Monroe Wall in 1967, having found it to be cytotoxic to KB cells in 1964.The structure was elucidated and published in 1971.
Even though it was an active compound showing modest activity against many leukimias, its modest yield from a relatively uncommon, slow growing tree and its insolubility in water made it an unlikely candidate. It proved to be strongly active in a mouse melanoma and as a result was selected for development in 1977.
In 1979, Susan Horwitz reported the mechanism of action for Taxol, specifically, in the formation of microtubules required for segregation of chromosomes. The renewed interest led to its clinical trials in 1984 and 1985. Clinically relevant responses were reported in 1989 and 1991 for ovarian and breast cancer. It is currently used along with its semi-synthetic analog docetaxel as single agents or in combination with other drugs such as cisplatin for ovarian, breast and non-small-cell lung cancer.
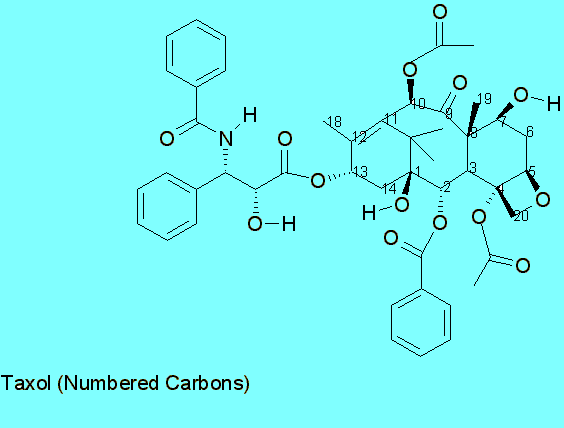
[1n]
[1a]
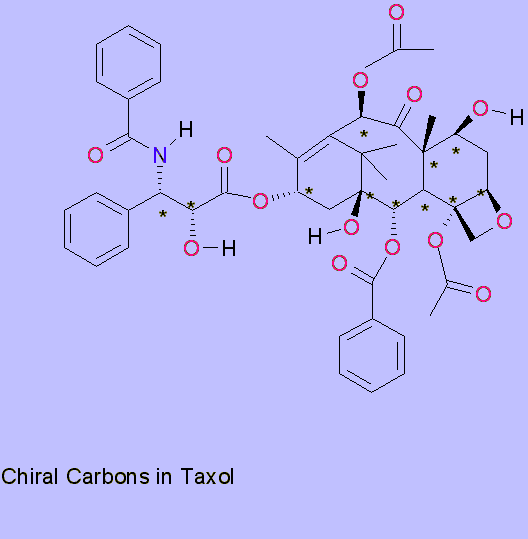
Bioactivity and functional groups5
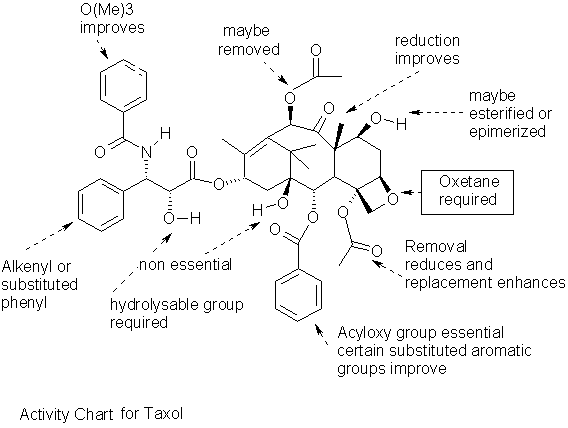
The study of activity by various scientists has led to a few general conclusions, such as the indispensable oxetane ring on C4 and C5. The open ring is inactive, however, a cyclopropyl derivative is equally as active. Some more active analogs such as one with carbonate instead of an acyl group at C4 are in clinical trials. Replacing oxygen in the ring with sulfur, presumably makes the ring too bulky, thus indicating size specificity.
Removing the benzoyl group from C2 or moving it to C1 seems to have a deactivating effect. Having a meta azido substiuent increases activity by an order of magnitude, in comparison to a para substitution. The deoxy analog for C1 proves a third less active. Linking the C14 and C1 with a carbonate yields a derivatives that is currently under development by Bayer.
The acyl phenyl group on the side chain may be replaced with an oxytrimethyl group for a more cytotoxic analog. The above mentioned derivative under development has this replacement as well as the removal of the other phenyl group from the side chain. The bioactivity of Taxol depends on the binding to the tubulin dimer, which disrupts the equilibrium between tubulin and microtubules, interrupting the cell cycle. Another mechanism which leads to hyper phosphorylation of a protein to which Taxol binds is also linked to the tubulin-assembly activity. Thus Taxol promoted tubulin assembly is thought to be biologically significant.
The above observations corroborate the findings observed in the structure, the oxetane ring is embedded in the tubulin, the phenyl rings seem to serve as a structural fit, and this key-lock fit excludes the north side of the molecule as drawn and the constituents at C7 to C10 are of minor consequence and may be replaced. The south side of the molecule has all the essential components for bioactivity, and as observed come closest to the atoms in the tubulin dimer.
Synthesis
It is probable that the amount of work done in synthesizing and testing may easily be underestimated, especially if one is not directly involved in the process. The various chiral centers and the large ring structure obviate a scheme for synthesis can at best be a trial among the myriad that actually led to its production. Among these are those found to involve fungus which produces trace amounts from plant cells, and of course those that use a partially biosynthetic scheme in which the baccatin III is obtained from plant extracts or fungal synthesis.
Retro synthesis( Step 1)
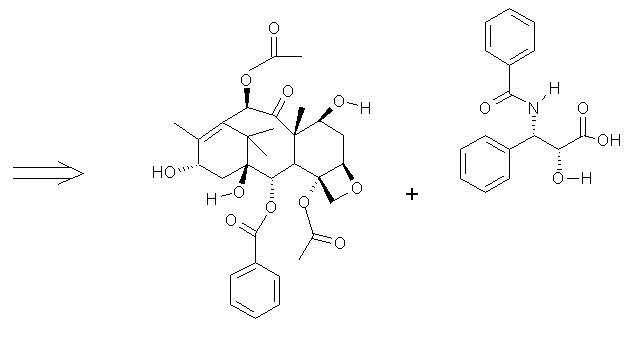
[2] ...........+............... [3]
The first and most obvious step is to remove the attachable side chain from the primary ring structure. Each of [2] and [3] could be synthesized separately, and esterified via acid catalyzation or reaction of the alcohol with acyl chloride. The presence of acyl groups on the ring may pose a problem which may be dealt with two ways, add the acyl groups after esterification or protect the acyl and hydroxyls at C1, C4, C7 & C10, with silyl groups such as TBDMS. Most of the literature has a similar approach, though some employ novel RCM4 with Ruthenium as catalyst and involve ring structures in both [2] & [3] simultaneously.
( Step 2) In the next step both[2] and [3] need be synthesized separately. As for [3],
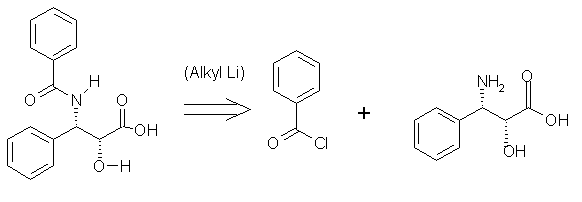
[3] .....>........ [4] .......+....... [5]
An acyl halide or even benzoic acid may be used to prepare [3], and oxidizing toluene with potassium permanganate, hydroxide ions and heat gives benzoic acid which may be treated with sulfonyl chloride to yield [4]. Alkyl Lithium may be used as a catalyst to join the amide to benzoyl chloride.The sketchy scheme shown in the following diagram betrays the lack of experience in synthesis. In the published schemes beta-lactams are used to produce the stereochemistry and ease of adding the ligand to C13.
More questions arose while trying to produce the two chiral carbons in the ligand than answers. Addition to double bonds can obviously be controlled as a syn or anti , but what is to stop the rotation about the single bond. The use of steric hinderance and ring addition seems to be the answer. Also one has to consider parallel reactions possible at other parts of the molecule. The choice of protecting groups is also important.
Review of various published syntheses 5
Most of the synthetic schemes concentrate on building the ABC tricycylic structure and four out of the six begin the construction with the six carbon A and C rings, forming the eight carbon B enclosure from these two later. The Kuwajima the Holton syntheses are discussed with considerable detail, while the others are mentioned.

Of these, there is only one that begins with a straight chain forming the B ring and then adding the C Ring, namely the Mukaiyama Synthesis.
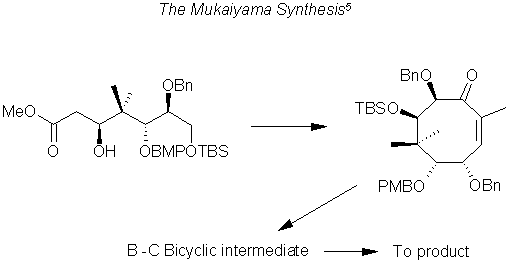
The Wender Synthesis begins with a Quadra cyclic structure leading to the A-B ring structure.
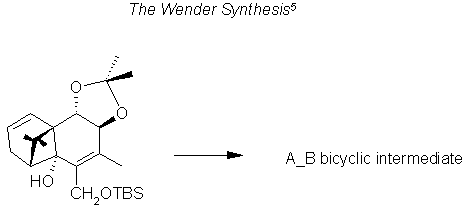
The RCM4 technique is employed to make a myriad of taxoids, and inversely the stereochemistry is simplified in a majority of syntheses by adding a group with a ring opening reaction conserving the stereo centers previously fabricated on the ring to be added.
Kuwajima Scheme for (-)- Taxol synthesis 1
Initial Fragments
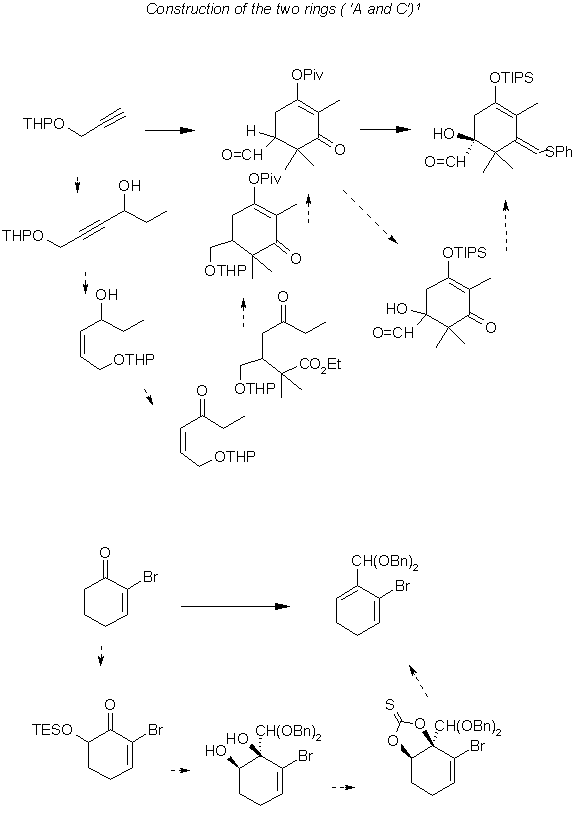
The two main problems in the synthesis of this compound are the construction of the tri cyclic structure and the specificity of the stereo centers. As in the case of others the C1 and C2 chirality is correct to begin with. For the construction of the A-Ring fragment Lithiated propargyl ether and propionaldehyde are mixed and by Lindlar reduction and oxidation gives the enone. A conjugate addition to the double bond by isobutyric enolate gives the keto ester which cyclizes with BuOK and after oxidation following removal of the protecting group yields the aldehyde shown as the intermediate product in the following diagram. Most of the above mentioned reactions occur below freezing point of water. (yield 50%)
In the next step Sharpless's asymmetric dihydroxylation yields the product shown as the pathway intermediate in the construction of the A ring but its isolation from the media was facilitated by converting it to its aminal by boiling with dimethylethylenediamine in benzene and then reconverting into its aldehyde via elution through silica gel.(yield 90%) (purity 98%)
The C-Ring fragment is prepared starting with 2-bromocyclohexenone forming predominantly the cis diol after oxidation and followed by addition of PhSCH(Li)OMe from the opposite side to the carbonyl group. The acetal added was changed to a dibenzyl group via dimethyl acetal. Conversion of the diol to thionocarbonate was followed by heating with diazaphospholidine yielding the cyclohexadiene shown as the product in the next illustration at the bottom of the page.
Each step of these reactions has been studied by others and the authors footnote their accomplishments which would be too time consuming for the present purpose to cite, however the source cited may lead to elucidation of each step. For this overview it should suffice to see the basic transformations and the relative efficiency or drawback of each operation.
The next step in this scheme is the combining of the A-ring and The C- ring fragments to form the beginning of the ABC structure. The advantage of using an aromatic C-Ring fragment is discussed in the next illustration its shown that the diene route is preferred for the difficulty in obtaining selective addition to the aromatic ring.
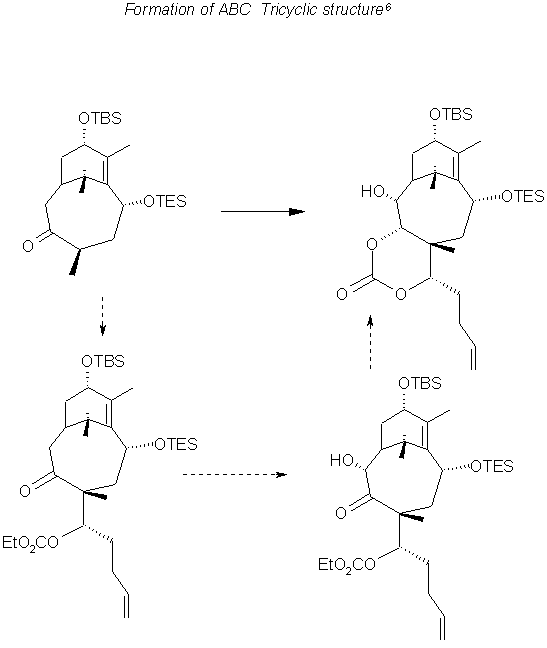
Skeleton Construction 1
A Grignard type reaction in which Magnesium alkoxide as the A Ring fragment and the lithiated C ring fragment are chelated to forms the starting material for the construction of the ABC skeleton. The C2 stereocenters seemed to be a pair of interconverting atropisomers. however the cyclized product had the desired conformation. Induced by a lewis acid subsequent to protection with Boron, the eight membered B ring contains the proper configuration fro the C9 and C10 carbons as well. The Boron protection was removed with pinacol and DMAP producing the resulting diol shown below.
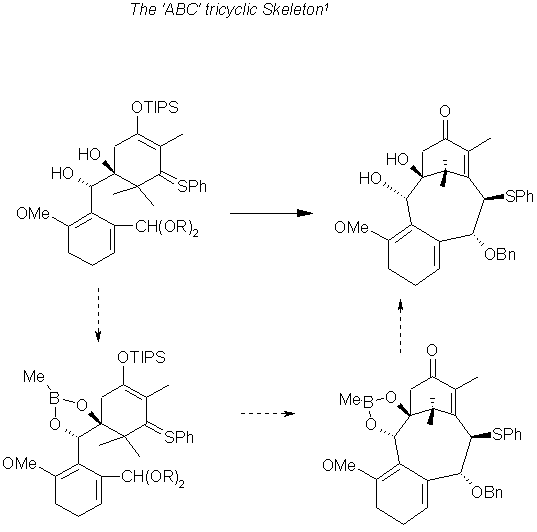
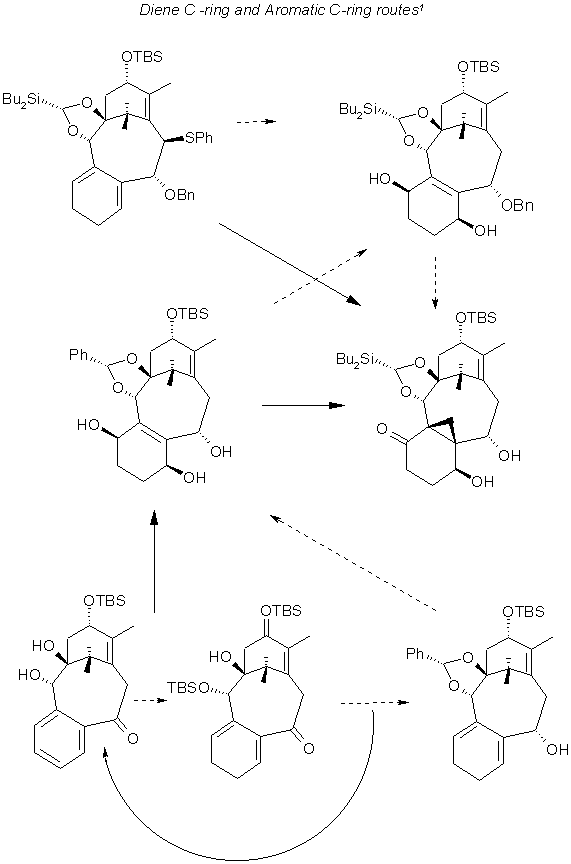
Comparison of aromatic and diene C-Ring routes 1
Protection of the diol from the previous step with Bu2Li, and TBSO yielded the starting materiel for the diene C-ring route. A succession of operations involving photoelectric oxygenation and subsequent reduction with Pd/C and H2 resulted in the triol which was protected and the benzyldione protected subsrtrate (C7, C9) cyclopropanated and upon acylation and cabonation yielded the product in the illustration (middle, right). Here the Bu2Li is preferred over Ph protection for C1, C2 for the cyclopropanation.
The aromatic C-ring posed problems in forming the starting material for the triol, as the intermediate (bottom,middle) had to be reoxidized as 88% of the product was an alcohol, rather than the ketone shown.. The same process of cyclopropanation yielded the final product. The failure of Birch reduction in the aromatic case, is attributed to the structure of the starting material (bottom, middle), the carbonyl is perpendicular to the benzene ring. The use of sterically hindered alcohols caused the benzene ring to be reduced but the yield was less than 50% and the remainder of the product was re-oxidized to the starting ketone.
Starting with an aromatic C-ring, 2-bromobenzaldehyde dibenzylacetal, yielded the ABC skeleton shown as the lower loop in the next illustration, after protection with PhCH(OMe)2, benzylidene, the product (middle, left) was obtained as in the previous case with catalytic photoelectric oxygenation.
If instead of TBSO as the protecting group at C2, SEM was employed during the reduction step, the bond cleavage was minimized but the removal of the protection group posed a difficulty at later stages. In the photoelectric oxidation step the catalyst in the diene route is TPP whereas in the aromatic case it is Rose Bengal and thiourea.
The cyclopropanation of the product(middle, left), benzyldene protected at C7 and C9 using Et2Zn/ClCH2I at the C3 C8 double bond failed because of the interaction of the oxygens at C2 and C4 with Zn. Successive treatment with BuLi and tBuSi(H)Cl yielded the starting material (top, left), for the formation of the cyclopropane at the double bond. The methylene transfer was also hindered by the conformations of the molecule the boat conformation of the 1, 3- dioxolane ring where the benzyllic hydrogen seems to prevent the approach of the reagent to the double bond. Interestingly, the subsequent cleavage of the propane ring to effect a methyl group at C8 is affected by the substituents at C13.
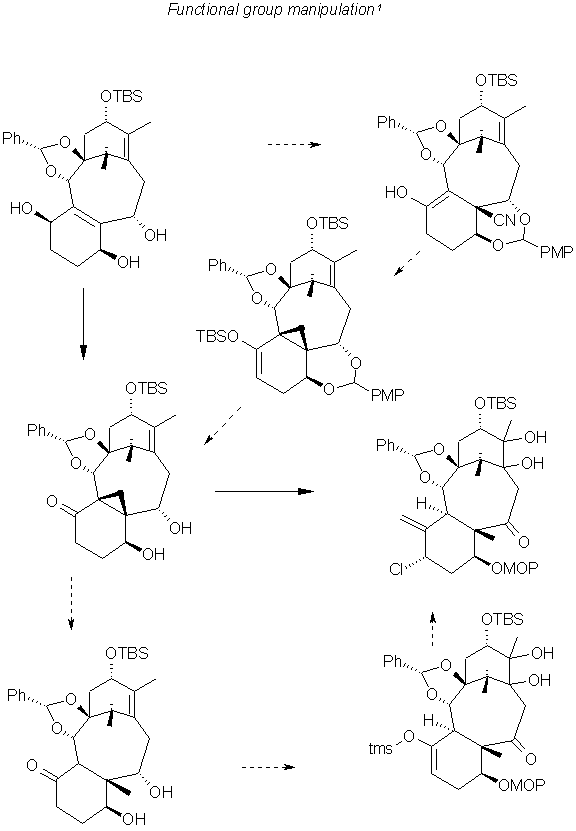
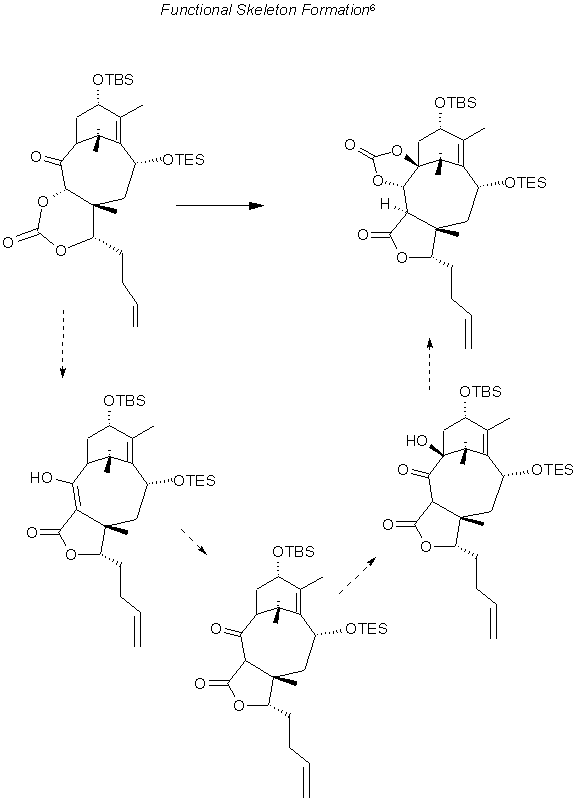
Addition of the Functional groups 1
The protecting groups had to be removed from C7,C9 and C1,C2 and even C13 for the cleavage of the cyclopropane, however the resulting triol was too unstable. So the TBSO was left on C13, the C7,C9 Ph was replaced by a carbonate and the SiBu2 removed and then replaced by a Ph on the C1,C2. the resulting C4 ketone(middle, right) then isomerized to give the triol with a methyl at C8. Of course for cleavage as mentioned earlier it was necessary to remove the protection from C13 as well.
Another method which involved less exchange of protecting groups, involved addition of the methyl, however, the methoxy group at C9 prevented addition at C8. Another protecting group was devised, with acetonide protecting C9,C10 a product methylated at C8 was observed. However introduction of this methyl group using carbanions or copper reagents to C7 substituted intermediates was to no avail. This led to the cyano addition noted in the following illustration (top, right) to (middle, right). The attempts to convert the cyano group to methyl using Wolff-Kishner reduction failed, and the displacement of the hydroxyl group from C19 methyl only resulted in the cyclopropane derivative.
The common synthetic intermediate (middle,right) was obtained from both the diene and the aromatic C-ring routes. The C7,C9 were initially protected as a boronic ester, C13 silylated and the boron group replaced with hydrogen peroxide. Selective oxidation at C9 with Dess-Martin perodinane, and subsequent protection of C7 OH the enolate at C4 ,C5 was cyclopropylated by treatment with KHMDS and with catalytiv amounts of Pd(ph3P)4 gave the chloride shown (middle, left) upon treatment with NCS in MeOH. OsO4 Oxidation resulted in producing the C11,C12 diol.
To effect hydroxylation of the exomethylene at C4, a functional group was introduced at C10 generating a C9,C10 enolate. The least hindered C4 group was observed to be when the B-ring was in a chair-boat and the C-ring in a chair conformation. Acetylation of the C10 hydroxyl group gave the B-ring chair-chair, C-ring boat conformer which readily isomerized to the previously mentioned and desired product(middle, bottom) in the next illustration. The oxetane ring formation was induced by heating with DBU.
Replacing the bulky protecting groups with less sterically hindered carbonate, effected the acetylation of C4 hydroxyl group remaining. The protecting groups were replaced and the final phase of the synthesis undertaken.
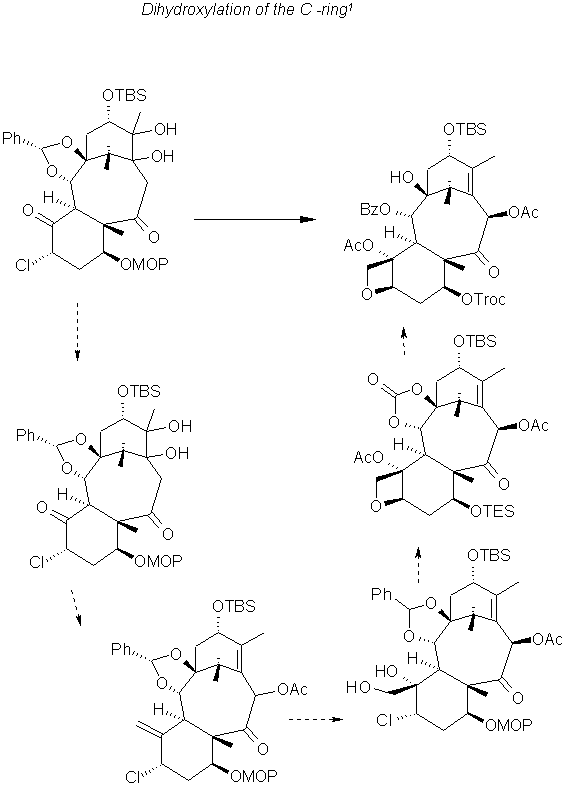
Final Steps 1
The acylation of C13 OH after removal of the protecting group by the lactam method using the four membered ring gave the final product for the (-)-taxol.
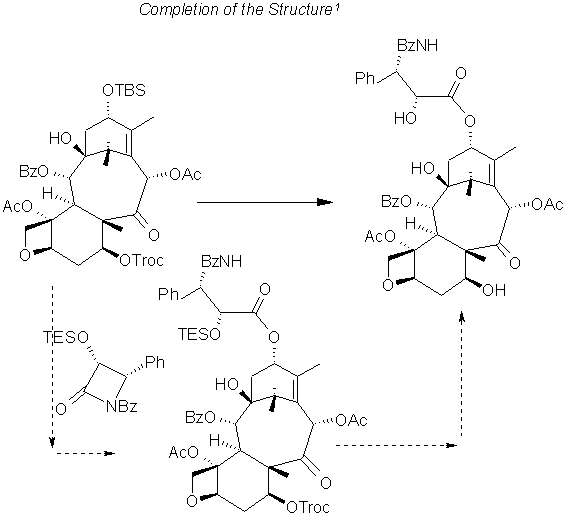
The Holton Synthesis 6
In their synthesis Holton et al start with an AB ring structure and emphasize the conformational control afforded by substituent effects on the 8 member B ring. The chair-boat conformation does not deprotonate at C8 so C10 silylation is used to effect 'epoxy alcohol fragmentation' to produce the intermediate (lower, left). The starting material is provided as a taxusin from camphor. C13 is also protected in this step.
Aldol condensation with 4- pentenal with magnesium enolate of the ketone gave hydroxy carbonate in chair-chair conformation. Reduction yielded the product (top, right).
Swern oxidation, gave the C2 ketone, it was expected that C3 would require a bulky substituent to change the conformation to a boat-chair so as to permit C1,C2 enolate generation, however, the ketone (top, right) in the next illustration underwent rearrangement while still in the chair-chair conformation allowing the removal of the C3 H. SmI2 reduction led to the (bottom, right) intermediate.
Attempts to produce and hydroxylate a dienolate from this were unsuccessful, but the cis and trans fused lactones (middle, bottom), the boat-chair conformation cis isomer predominant in yield(6:1) gave the product ( bottom, right). Apparently C1 hydrogen is removed first, even when C3 H is expected to be more acidic. The final product of the next illustration (top,right) is given by treatment with phosgene. This C1, C2, C3. C7 and C8 are functional as needed for taxol.
The steps in the synthesis are similar to the procedures used in the previous except C8 methylation is circumvented by the starting material and a rearrangement seems to fall into place sterospecifically. The Swern oxidation and pyridine and dichloromethane at low temperatures seem to be used regularly in both syntheses.
Toward completing the synthesis, oxidative cleavage by ozonolysis to the aldehyde followed by oxidation to the acid with esterification with diazomethane yields the starting material (top, left)for the last illustration for this synthesis. The first intermediate (top, right) is the result of the protocol developed by their lab. Decarbomethoxylation attempts yielded the starting material, protection with 2-methoxypropene, however, yielded better results(bottom, left).
The remainder of the synthesis is almost identical to the one described earlier. A bulky protecting group at C7, and the subsequent steps are very similar. The overall yield reported is 4-5%.
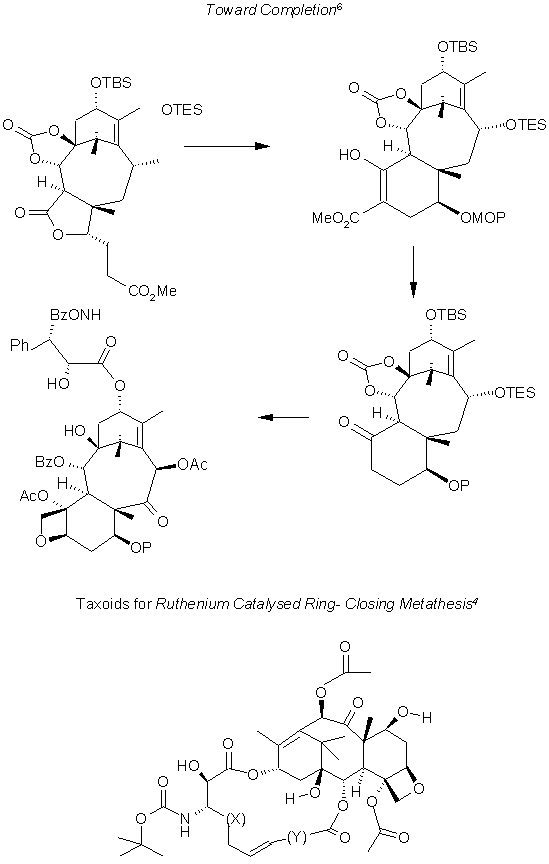
Summary
The RCM is employed to create a variety of Taxoids, with the same basic skeleton, and differing substituents at C2 and C13. Since the bioactivity of a large variety of chemicals can be tested this type of construction offers a valuable research tool. Among the few synthetic schemes studied, the pattern of using beta-lactams for adding the two chiral centers of the ligand is to be noted perhaps a well known tool among organic chemists. It is an art to manipulate conformations, selective substituent reactions by alternating between protecting groups and changing conformations with adding bulk at particular carbons. Binding a pair of substituents into position with protective carbonation or ring formation. It seems in actual practice a great deal of luck is depended upon. Indeed it is fascinating and creative work. I would have imagined myself to have bitten the proverbial mongoose's tail and shied from any study of organic chemistry au contraire I have renewed my zeal and interest for the subject alas experimentation is an absolute necessity as immersion into the field.References
- Hiroyuki Kusama, Ryoma Hara, Shigeru Kawahara, Toshiyuki Nishimori, Hajime Kashima, Nobuhito Nakamura, Koichiro Morihira and Isao Kuwajima, Enantioselective Total Synthesis of (-) - Taxol , J. Am. Chem. Soc. 2000, 122, 3811-3820
- Ryoma Hara, Takashi Furukawa, Hajime Kashima, Hiroyuki Kusama, Yoshiaki Horiguchi and Isao Kuwajima, Enantioselective Total Synthesis of (+) - Taxusin , J. Am. Chem. Soc. 1999, 121, 3072-3082
- David G. I. Kingston, Ashok G. Chaudhary, Mahendra D. Chordia, Milind Gharpure, A. A. Leslie Gunatilaka, Paul I. Higgs, John M. Rimoldi, Lakshman Samala and Prakash G. Jagtap, Synthesis and Biological Evaluation of 2- Acyl Analogues of Paclitaxel (Taxol) , J. Med. Chem. 1998, 41, 3715-3726
- Iwao Ojima, Songnian Lin, Tadashi Inoue, Michael L. Miller, Christopher P. Borella, Xudong Geng and John J. Walsh, Macrocycle Formation by Ring-Closing Metathesis. Application to the Syntheses of Novel Macrocyclic Taxoids , J. Am. Chem. Soc. 2000, 122, 5343-5353
- David G. I. Kingston, Taxol, a molecule for all seasons , Chem. Commun., 2001, 867-880
- Robert A. Holton, Carmen Somoza, Hyeong-Baik Kim, Feng Liang, Ronald J. Biediger, P. Douglas Boatman, Mitsuru Shindo, Chase C. Smith, Soekchan Kim, Hossain Nadizadeh, Yukio Suzuki, Chunlin Tao, Phong Vu, Suhan Tang, Pingsheng Zhang, Krishna K. Murti, Lisa N,. Gentile, and Jyanwei H. Liu, First Total Synthesis of Taxol , J. Am. Chem. Soc., 1994, 116, 1597-1600